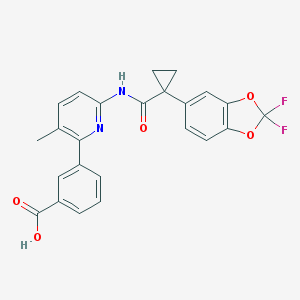
Maraviroc
In April 2012, Informa’s Scrip Insights published our book-length report, “Advances in the Discovery of Protein-Protein Interaction Modulators.” We also published a brief introduction to this report, highlighting the strategic importance of protein-protein interaction (PPI) modulators for the pharmaceutical industry, on the Biopharmconsortium Blog.
The report included a discussion on discovery and development of inhibitors of chemokine receptors. Chemokine receptors are members of the G-protein coupled receptor (GPCR) superfamily. GPCRs are seven-transmembrane (7TM) domain receptors (i.e. integral membrane proteins that have seven membrane-spanning domains). Compounds that target GPCRs represent the largest class of drugs produced by the pharmaceutical industry. However, in the vast majority of cases, these compounds target GPCRs that bind to natural small-molecule ligands.
Chemokine receptors, however, bind to small proteins, the chemokines. These proteins constitute a class of small cytokines that guide the migration of immune cells via chemotaxis. Chemokine receptors are thus a class of GPCRs that function by forming PPIs. Direct targeting of interactions between chemokines and their receptors (unlike targeting the interactions between small-molecule GPCR ligands and their receptors) thus involves all the difficulties of targeting other types of PPIs.
However, GPCRs–including chemokine receptors–appear to be especially susceptible to targeting via allosteric modulators. Allosteric sites lie outside the binding site for the protein’s natural ligand. However, modulators that bind to allosteric sites change the conformation of the protein in such a way that it affects the activity of the ligand binding site. (Direct GPCR modulators that bind to the same site as the GPCR’s natural ligands are known as orthosteric modulators.) In the case of chemokine receptors, researchers can in some cases discover small-molecule allosteric modulators that activate or inhibit binding of the receptor to its natural ligands. Discovery of such allosteric activators is much easier than discovery of direct PPI modulators.
Chemokines bind to sites that are located in the extracellular domains of their receptors. Allosteric sites on chemokine receptors, however, are typically located in transmembrane domains that are distinct from the chemokine binding sites. Small-molecule allosteric modulators that bind to these sites were discovered via fairly standard medicinal chemistry and high-throughput screening, sometimes augmented with structure-based drug design. This is in contrast to attempts to discover small molecule agents that directly inhibit binding of a chemokine to its receptor, which has so far been extremely challenging.
Our report describes several allosteric chemokine receptor modulators that are in clinical development, as well as the two agents that have reached the market. One of the marketed agents, plerixafor (AMD3100) (Genzyme’s Mozobil), is an inhibitor of the chemokine receptor CXCR4. It is used in combination with granulocyte colony-stimulating factor (G-CSF) to mobilize hematopoietic stem cells to the peripheral blood for autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma. The other agent, which is the focus of this blog post, is maraviroc (Pfizer’s Selzentry/Celsentri).
Maraviroc is a human immunodeficiency virus-1 (HIV-1) entry inhibitor. This compound is an antagonist of the CCR5 chemokine receptor. CCR5 is specific for the chemokines RANTES (Regulated on Activation, Normal T Expressed and Secreted) and macrophage inflammatory protein (MIP) 1α and 1β. In addition to being bound and activated by these chemokines, CCR5 is a coreceptor (together with CD4) for entry of the most common strain of HIV-1 into T cells. Thus maraviroc acts as an HIV entry inhibitor; this is the drug’s approved indication in the U.S. and in Europe. Maraviroc was discovered via a combination of high-throughput screening and optimization via standard medicinal chemistry.
New structural biology studies of the CCR5-maraviroc complex
Now comes a report in the 20 September 2013 issue of Science on the structure of the CCR5-maraviroc complex. This report was authored by a mainly Chinese group led by Beili Wu, Ph.D. (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai); researchers at the University of California at San Diego and the Scripps Research Institute, San Diego were also included in this collaboration. A companion Perspective in the same issue of Science was authored by P. J. Klasse, M.D., Ph.D. (Weill Cornell Medical College, Cornell University, New York, NY).
As described in the Perspective, the outer surface of the HIV-1 virus displays numerous envelope protein (Env) trimers, each including the outer gp120 subunit anchored in the viral membrane by gp41. When gp120 binds to the cell-surface receptor CD4, this enables interaction with a specific chemokine receptor, either CCR5 or CXCR4. Interaction with both CD4 and the chemokine receptor triggers complex sets of changes in the Env complex, eventually resulting in the fusion of the viral membrane and the cell membrane, and the entry of the virus particle into the host cell.
HIV-1 gp120 makes contact with CCR5 at several points. The interactions between CCR5 and the variable region of gp120 called V3 are especially important for the tropism of an HIV-1 strain, i.e., whether the virus is specific for CCR5 (the “R5 phenotype”) or CXCR4 (the “X4 phenotype”). In the case of R5-tropic viruses, the tip of the V3 region interacts with the second extracellular loop (ECL2) of CCR5, while the base of V3 interacts with the amino-terminal segment of CCR5. Modeling of the interactions between the V3 domain of gp120 of either R5 or X4-tropic viruses with CCR5 or CXCR4 explains coreceptor use, in terms of forming strong bonds or–conversely–weak bonds and steric hindrance.
Monogram Biosciences (South San Francisco, CA) has developed and markets the Trofile assay. This is a molecular assay designed to identify the R5, X4, or mixed tropism of a patient’s HIV strain. If a patient’s strain is R5-tropic, then treatment with maraviroc is appropriate. However, a patient’s HIV-1 strain may undergo a tropism switch, or may mutate in other ways to become resistant to maraviroc.
Dr Wu and her colleagues determined the high-resolution crystal structure of the complex between maraviroc and a solubilized engineered form of CCR5. This included determining the CCR5 binding pocket for maraviroc, which was determined both by Wu et al’s X-ray crystallography, and by site-directed mutagenesis (i.e., to determine amino acid residues that are critical for maraviroc binding) that had been published earlier by other researchers.
The structural studies of Dr. Wu and her colleagues show that the maraviroc-binding site is different from the recognition sites for gp120 and for chemokines, as expected for an allosteric inhibitor. The X-ray structure shows that maraviroc binding prevents the helix movements that are necessary for binding of g120 to induce the complex sequence of changes that result in fusion between the viral and cellular membranes. (These helix movements are also necessary for induction of signal transduction by binding of chemokines to CCR5.)
Structural studies of CXCR4 and its inhibitor binding sites
In addition to their structural studies of the CCR5-maraviroc complex, Dr. Wu and her colleagues also published structural studies of CXCR4 complexed with small-molecule and cyclic peptide inhibitors in Science in 2010. These inhibitors are IT1t, a drug-like orally-available isothiourea developed by Novartis, and CVX15, a 16-residue cyclic peptide that had been previously characterized as an HIV-inhibiting agent. IT1t and CVX15 bind to overlapping sites in CXCR4. Other researchers have found evidence that the binding site for plerixafor also overlaps with the IT1t binding site.
As discussed in Wu et al’s 2013 paper, CCR5 and CXCR4 have similar, but non-identical structures. The binding site for IT1t in CXCR4 is closer to the extracellular surface than is the maraviroc binding site in CCR5, which is deep within the CCR5 molecule. The entrance to the CXCR4 ligand-binding pocket is partially covered by CXC4’s N terminus and ECL2, but the CCR5 ligand-binding pocket is more open.
Mechanisms of CXCR4 and CCR5 inhibition, and implications for discovery of improved HIV entry inhibitors
The chemokine that specifically interacts with the CXCR4 receptor is known as CXCL12 or stromal cell-derived factor 1 (SDF-1). Researchers have proposed a hypothesis for how CXCL12 interacts with CXCR4; this hypothesis appears to be applicable to the interaction between other chemokines and their receptors as well. This hypothesis is know as the “two-step model” or the “two-site model” of chemokine-receptor activation. Under the two-site model, the core domain of a chemokine binds to a site on its receptor (known as the “chemokine recognition site 1” or “site 1”) defined by the receptor’s N-terminus and its ECLs. In the second step, the flexible N-terminus of the chemokine interacts with a second site (known as “chemokine recognition site 2” or “site 2” or the “activation domain”) deeper within the receptor, in transmembrane domains. This result in activation of the chemokine receptor and intracellular signaling.
Under the two-site model, CXCR4 inhibitors (e.g., IT1t, CVX15, and plerixafor), which bind to sites within the ECLs of CXCR4, are competitive inhibitors of binding of the core domain of CXCL12 to CXCR4 (i.e.., step 1 of chemokine/receptor interaction). They are thus orthosteric inhibitors of CXCR4. (This is contrary to the earlier assignment of plerixafor as an allosteric inhibitor of CXCR4.) The CCR5 ligand maraviroc, however, binds within a site within the transmembrane domains of CCR5, which overlaps with the activation domain of CCR5. Dr. Wu and her colleagues propose two alternative hypotheses: 1. Maraviroc may inhibit CCR5 activation by chemokines by blocking the second step of chemokine/chemokine receptor interaction, i.e., receptor activation. 2. Maraviroc may stabilize CCR5 in an inactive conformation. It is also possible that maraviroc inhibition of CCR5 may work via both mechanisms.
Dr. Wu and her colleagues further hypothesize that the interaction of HIV-1 gp120 with CCR5 (or CXCR4) may operate via similar mechanisms to the interaction of chemokines with their receptors. As we discussed earlier in this article, the base (or the stem region) of the gp120 V3 domain interacts with the amino-terminal segment of CCR5. The tip (or crown) of the V3 domain interacts with the ECL2 of CCR5, and–according to Dr. Wu and her colleagues–also with amino acid residues inside the ligand binding pocket; i.e., the activation site of CCR5. The HIV gp120 V3 domain may thus activate CCR5 via a similar mechanism to the two-step model utilized by chemokines.
Based on their structural biology studies, Dr. Wu and her colleagues have been building models of the CCR5-R5-V3 and CXC4-X4-V3 complexes, and are also planning to determine additional structures needed to fully understand the mechanisms of HIV-1 tropism. The researchers will utilize their studies in the discovery of improved, second-generation HIV entry inhibitors for both R5-tropic and X4-tropic strains of HIV-1.
The bigger picture
The 17 October 2013 issue of Nature contains a Supplement entitled “Chemistry Masterclass”. In that Supplement is an Outlook review entitled “Structure-led design”, by Nature Publishing Group Senior Editor Monica Hoyos Flight, Ph.D. The subject of this article is structure-based drug design of modulators of GPCRs.
This review outlines progress in determining GPCR structures, and in using this information for discovery of orthosteric and allosteric modulators of GPCRs.
According to the article, the number of solved GPCR structures has been increasing since 2008, largely due to the efforts of the Scripps GPCR Network, which was established in that year. Dr. Wu started her research on CXCR4 and CCR5 as a postdoctoral researcher in the laboratory of Raymond C. Stevens, Ph.D. at Scripps in 2007, and continues to be a member of the network. The network is a collaboration that involves over a dozen academic and industrial labs. Its goal has been to characterize at least 15 GPCRs by 2015; it has already solved 13.
Interestingly, among the solved GPCR structures are those for the corticotropin-releasing hormone receptor and the glucagon receptor. Both have peptide ligands, and thus work by forming PPIs.
One company mentioned in the article, Heptares Therapeutics (Welwyn Garden City, UK), specializes in discovering new medicines that targeting previously undruggable or challenging GPCRs. In addition to discovering small-molecule drugs, Heptares, working with monoclonal antibody (MAb) leaders such as MorphoSys and MedImmune, is working to discover MAbs that act as modulators of GPCRs. Among Heptares’ targets are several GPCRs with peptide ligands.
Meanwhile, Kyowa Hakko Kirin Co., Ltd. has developed the MAb drug mogamulizumab (trade name Poteligeo), which is approved in Japan for treatment of relapsed or refractory adult T-cell leukemia/lymphoma. Mogamulizumab targets CC chemokine receptor 4 (CCR4).
Thus, aided in part by structural biology, the discovery of novel drugs that target GPCRs–including those with protein or peptide targets such as chemokine receptors–continues to make progress.
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or of other issues that are important to your company, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.