Novel hypercholesterolemia drugs move toward FDA decisions
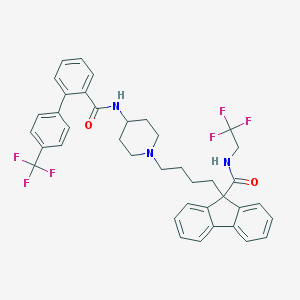
Lomitapide
Mid-October 2012 was a busy time for the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. On October 17, 2012, the panel voted 13-2 to recommend approval of Aegerion’s lomitapide for treatment of homozygous familial hypercholesterolemia. The next day, October 18, 2012, the same panel voted 9-6 to recommend approval of Isis/Sanofi/Genzyme’s mipomersen for the same condition.
Familial hypercholesterolemia (FH) is a rare genetic condition characterized by very high levels of low-density lipoprotein (LDL, or “bad cholesterol”), in the blood and early cardiovascular disease. Most patients with FH have mutations in either the LDL receptor (which functions to remove LDL from the circulation), or in apolipoprotein B (ApoB) (the protein moiety of LDL, which binds to the LDL receptor).
Patients who are heterozygous for an FH mutation (but have one normal copy of the affected gene) may have premature cardiovascular disease in their thirties. Patients who are homozygous for an FH mutation may have severe cardiovascular disease in childhood. Heterozygous FH is a common genetic disease, which is inherited in an autosomal dominant pattern, and occurs in one out of 500 people. Homozygous FH, however, occurs in about 1 in a million births. Homozygous FH thus qualifies as a “rare disease”.
Physicians generally treat heterozygous FH with statins, bile acid sequestrants or other lipid-lowering agents that lower cholesterol levels. Homozygous FH often does not respond to these drugs. It may require chronic treatment via LDL apheresis (removal of LDL in a method similar to dialysis) and in some cases liver transplantation.
Aegerion (Cambridge, MA), the developer of lomitapide, is a publicly-traded biotech company that seeks to “change the way that rare, genetic lipid disorders are treated”. It is currently focused on the development of lomitapide, a small-molecule compound (pictured above).
Lomitapide inhibits the microsomal triglyceride transfer protein (MTTP) which is necessary for very low-density lipoprotein (VLDL) assembly and secretion in the liver. A 2007 article in the New England Journal of Medicine (NEJM) concluded that inhibition of MTTP by lomitapide (then known as BMS-201038) resulted in the reduction of LDL cholesterol levels in patients with homozygous FH. BMS-201038/lomitapide was originally developed by Bristol-Myers Squibb (BMS), donated to the University of Pennsylvania in 2003 and licensed to Aegerion in 2006. BMS had abandoned development of the compound after early Phase 1 and Phase 2 trials had found increases in heptatic fat content and gastrointestinal disturbances. The NEJM study (conducted by Penn researchers in collaboration with other academic researchers and with BMS) also found that therapy with the compound was associated with elevated liver aminotransferase levels and hepatic fat accumulation.
78-week data from Aegerion’s pivotal Phase 3 study of lomitapide in adults patients with homozygous FH were published in the online version of The Lancet on November 2, 2012.
Mipomersen (which will be called Kynamro if and when it is commercialized) is an antisense oligonucleotide that targets the messenger RNA for apolipoprotein B. We discussed mipomersen in our August 21, 2009 blog article on oligonucleotide therapeutics. Mipomersen represents the most advanced oligonucleotide drug in development that is capable of systemic delivery. (The only two marketed oligonucleotide drugs both treat ophthalmologic diseases and are delivered locally.) Mipomersen targets the liver, without the need for a delivery vehicle. Thus mipomersen–potentially the first systemically-delivered oligonucleotide drug to reach the market–represents the “great hope” for proof-of-concept for oligonucleotide drugs, including antisense and RNAi-based drugs.
Patients treated with mipomersen, as with lomitapide, exhibit liver-related adverse effects, especially hepatic fat accumulation and elevated liver aminotransferase levels. Moreover, unlike lomitapide, which is an orally-delivered compound, mipomersen, which is delivered via subcutaneous injection, can cause injection site reactions and flu-like symptoms. Moreoever, mipomersen has a much longer half-life than lomitapide (30 days versus 20 hours).
Industry commentators, and well as the FDA Advisory Committee, generally favor lomitapide over mipomersen, because lomitapide appears to be the more efficacious drug in lowering LDL-cholesterol, and also because lomitapide is an oral drug. However, most of the FDA panelists, as well as other industry commentators believe that not all patients with homozygous FH would be likely to benefit from only one drug. Thus having two alternative drugs may well be better in treating this disease.
Both lomitapide and mipomersen have potentially serious adverse effects. A finding of elevated liver aminotransferase levels is enough to stop development of most drugs. However, the FDA and its Advisory Panel believe that a risk evaluation and mitigation strategy (REMS) would support appropriate use of these drugs in patients with homozygous FH, because of their life threatening disease, and because they have limited therapeutic options. Both Aegerion and Genzyme are proposing that their compounds be approved with REMS programs, including an education program for physicians and active monitoring of patients. The REMS program would also include monitoring to ensure that only adult homozygous FH patients would be treated with the drugs. However, Aegerion plans to conduct clinical trials of the use of lomitapide in pediatric homozygous FH patients, as well as patients with another rare disease, familial chylomicronemia. Genzyme has already tested mipomersen in a small number of pediatric patients.
Companies developing therapeutics for rare diseases whose mechanisms are related to those of more common diseases often attempt to first get their drugs approved for the rare disease, and then perform additional clinical trials to expand the drug’s indications to larger populations. We discussed this strategy in an earlier article on this blog. Homozygous FH is mechanistically related to not only heterozygous FH, but also to cases of severe hypercholesterolemia that are poorly controlled by statins. Both companies have shown interest in treating patients with homozygous FH and severe hypercholesterolemia, since they have preformed clinical trials that included patients with these conditions. However, the adverse effects of these drugs may limit their use to homozygous FH, at least in the near future.
Aegerion intends to market lomitapide on its own, and is ramping up its marketing and sales organization in anticipation of approval. Mipomersen, if approved, would have the benefit of the Sanofi marketing organization behind it. However, industry commentators expect lomitapide to have a large advantage over mipomersen, if both are approved. That is because of the greater efficacy of lomitapide, its oral dosing, and other factors related to injection site reactions for mipomersen and the half-lives of the compounds.
We await FDA action in the next several weeks on the approval of lomitapide and mipomersen.
Meanwhile, researchers and companies are working on potential drugs for severe hypercholesterolemia that act via an entirely different mechanism–PCSK9 (proprotein convertase subtilisin/kexin 9) inhibition. These drugs are in an earlier stage of development than lomitapide and mipomersen. However, they might eventually provide strong competition to these drugs, or replace them altogether.
For oligonucleotide drug developers and enthusiasts, the case of mipomersen–considered the “great hope” for proof-of-concept for oligonucleotide drugs by many in the field–provides several lessons. 1. At the end of the day, oligonucleotide drugs must meet the same standards of safety and efficacy as other drugs. 2. Oligonucleotide drugs may encounter competition from drugs in other classes, such as small molecules or monoclonal antibodies.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.
