A new wave of small-molecule disease-modifying drugs for cystic fibrosis
Ivacaftor
In our January 24, 2013 article on this blog, we discussed the cases of two genetic diseases, sickle cell disease (SCD) and cystic fibrosis (CF). In both cases, the genetic cause of the disease was identified decades ago. However, no disease-modifying drugs for SCD have yet been developed.
In the case of CF, small-molecule disease-modifying drugs have only recently entered the pipeline. In one case, such a drug–ivacaftor (Vertex’ Kalydeco), was approved both in the U.S. and in Europe in 2012.
In this article, we discuss the development of small-molecule drugs for CF.
Cystic fibrosis
As we discussed in our earlier article, CF causes a number of symptoms, which affect the skin, the lungs and sinuses, and the digestive, endocrine, and reproductive systems. Notably, people with CF accumulate thick, sticky mucus in the lungs, resulting in clogging of the airways due to mucus build-up. This leads to inflammation and bacterial infections. Ultimately, lung transplantation is often necessary as the disease worsens. With proper management, patients can live into their late 30s or 40s.
The affected gene in CF and the most common mutation that causes the disease (called ΔF508 or F508del) were identified by Francis S Collins, M.D., Ph.D. (then at the Howard Hughes Medical Institute and Departments of Internal Medicine and Human Genetics, University of Michigan, Ann Arbor, MI) and his colleagues in 1989. (Dr. Collins was subsequently the leader of the publicly-funded Human Genome Project and is now the Director of the National Institutes of Health, Bethesda, MD.)
The gene that is affected in cystic fibrosis encodes a protein known as the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR regulates the movement of chloride and sodium ions across epithelial membranes, including the epithelia of lung alveoli. CF is an autosomal recessive disease, which is most common in Caucasians; one in 2000–3000 newborns in the European Union is found to be affected by CF. ΔF508 is a deletion of three nucleotides that causes the loss of the amino acid phenylalanine at position 508 of the CFTR protein. The ΔF508 mutation accounts for approximately two-thirds of CF cases worldwide and 90% of cases in the United States. However, there are over 1500 other mutations that can cause CF.
CFTR is an ion channel–specifically a chloride channel. Ion channels constitute an important class of drug targets, which are targeted by numerous currently marketed drugs, e.g., calcium channel blockers such as amlodipine (Pfizer’s Norvasc; generics) and diltiazem (Valeant’s Cardizem; generics). These compounds were mainly developed empirically by traditional pharmacology before knowing anything about the molecular nature of their targets.
However, discovery of novel ion channel modulators via modern molecular methods has proven to be challenging, mainly because of the difficulty in developing assays suitable for drug screening. In addition, development of suitable assays for assaying chloride channel function has lagged behind the development of assays for the function of cation channels (e.g., sodium and calcium channels).
Moreover the most common CFTR mutation that causes CF, ΔF508, results in defective cellular processing, and the mutant CTFR protein is retained in the endoplasmic reticulum. In the case of some other mutant forms of CTFR (accounting for a small percentage of CF patients), the mutant proteins reach the cell membrane, but are ineffective in chloride-channel function.
Vertex’ program for the development of small molecule CF drugs
Efforts aimed at the discovery of small-molecule drugs for CF began in 1998, when the nonprofit Cystic Fibrosis Foundation (CFF) established its Therapeutics Development Program. This included a drug discovery unit, through which CFF would support both academic and industrial research. An early recipient of CFF funding via this program was a small biotech company, Aurora Biosciences (San Diego, CA). Aurora had developed technology for ultra-high-throughput screening in cellular assays, which they were applying to the discovery of small-molecule drugs for CF. In 2001, Vertex Pharmaceuticals (Cambridge, MA) acquired Aurora. Vertex then incorporated Aurora’s technology into its drug discovery programs, including its CF program. Vertex’ CF program received continuing support from CFF.
Vertex researchers screened thousands of drug-like and lead-like synthetic compounds in recombinant mouse cells expressing mutant human CFTR. Positive hits that met criteria for developability were further tested in a rat epithelial cell line that expressed the mutant CFTR. Compounds selected for further study were also tested for rescue of CFTR activity in cultured primary human lung airway epithelial cells from CF patients, which expressed the same mutant CFTRs studied in the recombinant cell assays. Performing the latter studies required isolating the epithelial cells from lung tissue of CF patients. The thick mucus found in CF lungs made this isolation very challenging. According to Vertex researcher and project head Fred Van Goor, researchers had to use tweezers to extract the mucus, in order to isolate the cells. It reportedly took all of 2003 to develop cellular assays (both in primary and recombinant cells) to conduct the drug discovery studies.
Vertex’ high-throughput screening studies resulted in the identifications of two types of lead compounds:
- CFTR potentiators, which potentiate the chloride channel activity of mutant CFTR molecules at the cell surface;
- CFTR correctors, which partially correct the folding and/or trafficking defect of such mutant CFTRs as ΔF508, thus facilitating exit from the endoplasmic reticulum and deposition of a portion of the mutant CFTR in the cell membrane.
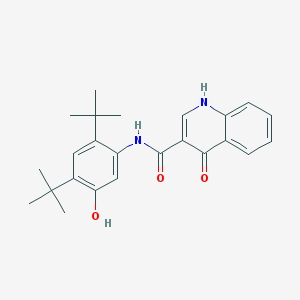
Vertex’ ivacaftor, a CFTR potentiator
The Vertex screening studies discussed in the previous section, published in 2006, resulted in clinical candidates in both the potentiator and corrector classes. The company pursued development of one potentiator compound, ivacaftor (formerly known as VX-770) (Vertex’ Kalydeco). Ivacaftor is indicated only in patients with the G551D (Gly551Asp) mutation in CFTR, which only accounts for around 4% of CF patients.
Ivacaftor was discovered via high-throughput screening as described in the previous section, followed by lead optimization. The compound increased chloride channel function both in recombinant cells carrying mutant CFTR, and in cultured primary human lung airway epithelial cells from CF patients. Ivacaftor was found to be efficacious in opening both CFTR G551D and CFTR ΔF508 present in the cell membranes of recombinant cells. However, because of the retention of CFTR ΔF508 in the endoplasmic reticulum in human lung airway epithelial cells, this compound is not efficacious in treating CF patients carrying this mutation. The lack of efficacy in patients homozygous for CFTR ΔF508 was confirmed in a subsequent clinical trial.
On February 23, 2011, that a Phase 3 trial of ivacaftor (then called VX-770) showed marked improvement in lung function in CF patients carrying the CFTR G551D mutation. Treated patients also had significant weight gain, showed reduced sweat chloride (a CF biomarker), and were less likely to have a pulmonary exacerbation. The results of this Phase 3 trial were published in the New England Journal of Medicine. Also in 2011, Vertex submitted a New Drug Application (NDA) for ivacaftor. In January 2012, the FDA approved ivacaftor for treatment of CF patients carrying the CFTR G551D mutation. In July 2012, Vertex received European approval for this drug.
Vertex’ lumacaftor (VX-809) and VX-661, CFTR correctors
Vertex is currently developing the CFTR corrector lumacaftor (VX-809). The company has completed Phase 2 studies of a combination of ivacaftor and lumacaftor/VX-809 in CF patients who are homozygous for the CFTR ΔF508 mutation. It is now planning pivotal phase 3 trials of the combination therapy in this patient population. The rationale for the combination treatment is that VX-809 potentates the deposition of CFTR ΔF508 in the cell membrane, and invacaftor potentiates the function of cell-surface CFTR ΔF508.
Vertex is also conducting Phase 2 trials of another CTFR corrector, VX-661, alone and in combination with ivacaftor/VX-770 in CF patients homozygous for CFTR ΔF508.
The Cystic Fibrosis Foundation’s collaboration with Pfizer
The CFF has also been collaborating with Pfizer to discover drugs to treat patients carrying the the CFTR ΔF508 mutation. This collaboration began after the 2010 acquisition by Pfizer of FoldRX (Cambridge, MA). In November 2012, the CFF and Pfizer expanded their collaboration.
The Pfizer/CFF collaboration builds on FoldRx’s cystic fibrosis research program in collaboration with the CFF, which started in 2007. FoldRX (now a wholly-owned subsidiary of Pfizer) specializes in discovering and developing drugs to treat diseases of protein misfolding. The correction of protein misfolding clearly applies to CFTR ΔF508 protein.
Under the expanded six-year CFF/Pfizer collaboration, the CFF will invest up to $58 million to support and accelerate the discovery and development of disease-modifying therapies for CFTR ΔF508 CF. The goal of the collaboration is to advance one or more drug candidates into the clinic by the end of the six-year period. The CFF will provide scientific as well as financial support to help advance this discovery program.
Ataluren, for treatment of patients with CFTR nonsense mutations
Ataluren (formerly known as PTC124), is being developed by PTC Therapeutics for various genetic diseases caused by nonsense mutations in critical genes. The drug is currently being investigated for use in patients with nonsense mutation Duchenne/Becker muscular dystrophy (DBMD) and cystic fibrosis (CF). PTC Therapeutics’ efforts in CF are supported by a grant from the CFF.
Ribosomes normally translate messenger RNAs (mRNAs) into protein until arriving at a normal stop codon in the mRNA, at which point the ribosome stops translation, resulting in a functional protein. Nonsense mutations, however, create a premature stop signal in the mRNA coding sequence. This causes the ribosome to stop translation before a functioning protein is generated, creating a truncated, nonfunctional protein. This can result in disease.
Ataluren is designed to allow the ribosome to ignore the premature stop signal and continue translation of the mRNA, resulting in formation of a functioning protein. Ataluren does not cause the ribosome to read through the normal stop signal.
The results of clinical trials of ataluren in pediatric (Phase 2a) and adult (Phase 2) patients with nonsense-mutation CF showed that the drug resulted in production of functional CFTR protein and statistically significant improvements in CFTR chloride channel function. Ataluren treatment was also associated with significant reductions in cough frequency and trends toward improvement in pulmonary function tests.
Conclusions
As we discussed in our January 24, 2013 article on this blog, the 1989 identification of the genetic cause of CF did not immediately lead to the development of disease-modifying drugs. Bottlenecks in the pathway from genetic research to small-molecule drugs included understanding the different ways (e.g., deficiencies in chloride channel function, deficiencies in protein processing, blockages in protein translation due to nonsense mutations) in which the many mutations that can cause CF act, and the need to develop effective assays for use in drug discovery.
The 2012 approval of the CFTR potentiator ivacaftor (Vertex’ Kalydeco) in the U.S. and Europe represents a real milestone in CF drug development. Vertex and the CFF should be congratulated on their breakthrough CF R&D program, which required the willingness to pursue a long pathway to development.
Other compounds that target CFTR are in Phase 2 development. All indications suggest that treatment for CF will represent a case of “personalized medicine”, as befits a disease that is caused by multiple mutations that act at different levels of protein synthesis, processing, and function.
As is typical for personalized medicines that target rare diseases, Kalydeco is expensive. The drug reportedly costs upwards of $294,000 for a year’s supply. Vertex says that it will supply Kalydeco free to U.S. patients with no insurance and a household income of under $150,000.
With the interest of pharmaceutical and biotechnology companies in developing targeted therapies and therapies for rare diseases, the story of the development of small-molecule drugs for CF represents an important case study in drug discovery and development in the 2010s. , the emphasis on targeted drugs and rare diseases has also resulted in the the recent increase in FDA drug approvals; the agency approved 39 new drugs in 2012, which represents a 16-year high.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.
