FDA publishes Draft Guidance on developing drugs for early stages of Alzheimer’s disease
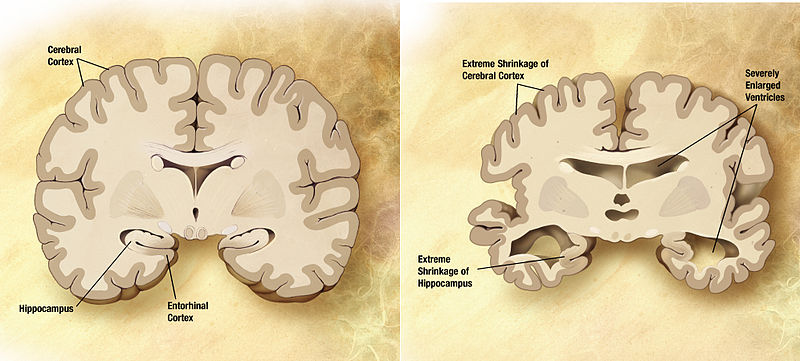
Normal and Alzheimer’s brains compared.
Once again, approaches to improving clinical trials for candidate disease-modifying drugs for Alzheimer’s disease (AD) are in the news. On February 7, 2013, the FDA issued a Draft Guidance for Industry entitled “Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease”.
This document has been distributed for comment purposes only, and the FDA is seeking public comment on the draft guidance for 60 days.
The wording of the Draft Guidance illustrates the extreme difficulty of defining populations with pre-AD or very early-stage AD, and of demonstrating the efficacy of a drug in ameliorating early-stage disease, and/or in preventing its progression to later-stage disease. The document states that the FDA is “open to considering the argument that a positive biomarker result (generally included as a secondary outcome measure in a trial) in combination with a positive finding on a primary clinical outcome measure may support a claim of disease modification in AD.”
However, there is currently no evidence-based consensus as to which biomarkers might be appropriate to support clinical findings in trials in early AD. Moreover, in “pre-AD” or very early-stage AD (i.e., before the onset of overt dementia) mild disease-related impairments are extremely challenging to assess accurately. Thus both measuring clinical outcomes and assessment via biomarkers in very early-stage AD are fraught with difficulty, making determination of drug efficacy extremely difficult. The FDA thus appears to be seeking guidance from industry and from the academic community on how these knotty problems might be solved.
The move toward conducting clinical trials in early-stage AD patients
By issuing the Draft Guidance, the FDA adds its voice to that of an ever-increasing segment of the scientific community that calls for a new focus on conducting clinical trials in early-stage AD. We discussed this trend in our August 19, 2012 and August 28, 2012 articles on the Biopharmconsortium Blog.
As we discussed, this trend is driven in part by the Phase 3 failures of Pfizer/Janssen’s bapineuzumab and Lilly’s solanezumab in 2012. Now–in February 2013–Russell Katz, M.D. (director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research) says, “The scientific community and the FDA believe that it is critical to identify and study patients with very early Alzheimer’s disease before there is too much irreversible injury to the brain. It is in this population that most researchers believe that new drugs have the best chance of providing meaningful benefit to patients.” In line with this statement, the FDA refused to entertain Lilly’s secondary analysis of early stage patients in the solanezumab study that we discussed in our August 28, 2012 blog article. Instead, the FDA mandated that Lilly conduct a new Phase 3 trial that will exclude the moderate-stage patients who hadn’t responded, and focus only on early-stage patients.
Recent news on clinical trials in early-stage AD
Despite the difficulties highlighted in the Draft Guidance in conducting clinical trials in early-stage AD patients, three research groups are actually conducting such trials. We outlined these studies in our August 28, 2012 blog article, and discussed one of these studies, the one begin carried out by Genentech, in greater detail in our August 19 2012 article.
The three studies are:
- Roche/Genentech’s Phase 2a trial of its its anti-amyloid MAb crenezumab, in presymptomatic members of a large Colombian kindred who harbor a mutation in presenilin 1 (PS1) that causes dominant early−onset familial AD.
- Studies conducted in conjunction with the Dominantly Inherited Alzheimer Network (DIAN), a consortium led by researchers at Washington University School of Medicine (St. Louis, MO). This study will include people with mutations in any of the three genes linked to early-stage, dominantly-inherited AD–PS1, PS2, and amyloid precursor protein (APP). Initial studies focused on changes in biomarkers and in cognitive ability as a function of expected age of AD onset in people with these mutations. These included changes in concentrations of amyloid-β1–42 (Aβ42) in cerebrospinal fluid (CSF), and amyloid accumulation in the brain. In the first stage of the actual trial, three drugs (which have not yet been selected) will be tested in this population, and changes in biomarkers and cognitive performance will be followed.
- The Anti-Amyloid Treatment of Asymptomatic Alzheimer’s (A4) trial, will involve treating adults without mutations in any of the above three genes, whose brain scans show signs of amyloid accumulation. A4 is thus designed to study prevention of sporadic AD (by far the most common form of the disease). It will enroll 500 people age 70 or older who test positive on a scan of amyloid accumulation in the brain. (This is in contrast to the two trials in subjects with gene mutations, who are typically in their 30s or 40s.) A4 will also have a control arm of 500 amyloid-negative subjects. Amyloid-positive and control subjects will be entered into a three-year double-blind clinical trial that will look at changes in cognition with drug treatment. The A4 researchers [led by Reisa Sperling, Brigham and Women’s Hospital/Harvard University (Boston, MA), and Paul Aisen, University of California, San Diego] planned to select a drug for testing by December 2012.
Now there is more recent news on two of these trials.
1. On December 13, 2012, the Los Angeles Times reported that Genentech and its collaborators [affiliated with the University of Antioquia medical school (Medellin, Colombia), the University of California at Los Angeles (UCLA), and the Banner Alzheimer’s Institute (Phoenix, AZ)] will begin their $100 million clinical trial of crenezumab with 100 Colombians who carry the PS1 mutation in the spring of 2013. Genentech is contributing $65 million of the study’s $100-million cost. The NIH and the Banner Alzheimer’s Institute (Phoenix, AZ) are financing the remainder.
This story was also reported on December 14, 2012 by Fierce Biotech.
The design of the trial calls for 100 additional patients in Colombia with the same Alzheimer’s-related gene to receive a placebo, and an equal number of other at-risk patients without the gene to take crenezumab. A branch of the trial will include U.S. patients as well. A “branch study” will also be conducted at UCLA, where researchers have discovered a similar genetic disposition among members of an extended family from Jalisco, Mexico. Some 30 individuals from this family who have immigrated to Southern California could participate. Around 150 other U.S. patients with similar mutations will also participate in the trial.
The trial is designed to provide evidence that targeting amyloid with crenezumab at an early stage or even before patients show signs of dementia can have a positive effect on the course of disease.
2. On January 18, 2013, Fierce Biotech reported that the researchers conducting the A4 study have chosen Lilly’s solanezumab as as the first therapeutic drug candidate to be evaluated in the trial. The A4 trial’s principal investigator, Reisa Sperling said that the researchers chose solanezumab (after considering a number of anti-amyloid drugs) because the compound has a good safety profile, and appeared to show a modest clinical benefit in the mild AD patients in Lilly’s Phase 3 trial. The A4 researchers’ confidence in solanezumab grew when this was confirmed via an independent academic analysis by the Alzheimer’s Disease Cooperative Study (ADCS), a consortium of academic Alzheimer’s disease clinical trial centers. The ADCS, which was established by NIH, will help facilitate the A4 trial.
The A4 researchers hope that starting treatment with solanezumab before symptoms are present, as well as treating for a longer period of time, will slow cognitive decline and ultimately prevent AD dementia.
After the failure of solanezumab in Lilly’s own Phase 3 studies, and the FDA’s rebuff of the company’s secondary analysis of early stage patients, the A4 study’s choice of solanezumab gives the drug a new lease on life. Meanwhile, Lilly will be continuing its own clinical trial program for solanezumab.
Conclusions
The three clinical trials discussed in this article should allow the scientific and medical community to answer the question as to whether treating patients with pre-AD or very early-stage AD with anti-amyloid MAb drugs can have a positive effect on the course of the disease, and slow or prevent cognitive decline. The studies may also help the scientific and medical community, and the FDA, with issues of evaluation of biomarkers and clinical outcome measures in determining disease prognosis and the efficacy of drug treatments. Given the large size and rapid growth of the at-risk population, finding safe and efficacious disease-modifying preventives and treatments for AD is of increasing urgency.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.