![]()
Haberman Associates now has a new website, which is available at https://biopharmconsortium.com. Our new site has an attractive, up-to-date look and feel, and also contains updated news and other updated content.
We have also fully integrated our Biopharmconsortium Blog into our new site. Our blog resides at https://biopharmconsortium.com/blog. There has been a hiatus in posting on our blog recently, due to finishing construction of our new site. Thus the last article on our blog before completion of the website was posted on April 5, 2013. However, we posted a new article on May 29, 2013, and intend to publish blog articles on a more regular basis.
The integration of the Biopharmconsortium Blog into our website emphasizes that it is the blog for our consulting group, not an independent entity or a journalistic blog. Despite the diversity of subjects covered by the blog, the focus is on effective drug discovery and development, and on company strategies designed to facilitate effective new product development.
For more information on our consulting group, Haberman Associates, see our brief December 16, 2012 blog article, entitled What Is Haberman Associates? Better yet, you may explore our website for a more complete picture of our consulting group.
We hope that the diverse community of our readers will benefit from the discussions on our blog. We also hope that those of you in life science companies will get a feeling for how we approach issues in drug discovery and development and company strategies.
However, even the best articles or books on how to solve key industry problems will not solve these problems on their own. Companies need to develop company-specific solutions and to implement them. Haberman Associates consulting may enable your company to formulate and implement the solutions you need to improve your productivity.
If you are in a life sciences firm, and have questions about Haberman Associates, or wish to send us a consulting inquiry or to commission us to write a report for publication, please telephone or e-mail us.

Sir2, the yeast homologue of SIRT1
The Biopharmconsortium Blog has from time to time been following novel developments in anti-aging medicine, including attempts to develop activators of sirtuins. However, we have not had an article on sirtuins since December 1, 2010. At that time, we reported on the discontinuation by GlaxoSmithKline (GSK) of its lead sirtuin activator, SRT501, a proprietary formulation of the natural product resveratrol (which is found in red wine).
Sirtuins are nicotinamide adenine dinucleotide (NAD+)–dependent protein deacetylases, which have been implicated in control of lifespan in yeast, the nematode Caenorhabditis elegans, and the fruit fly Drosophila. Mammalian sirtuins have been implicated (via animal model studies) in protection against aging-related diseases such as metabolic and cardiovascular diseases, neurodegeneration, and cancer.
As we discussed in our December 1, 2010 article, GSK acquired the sirtuin-pathway specialty company Sirtris (Cambridge, MA) for $720 million in June 2008. This gave GSK ownership of Sirtris’ sirtuin modulator drugs. As stated in that article, although GSK discontinued development of SRT501, it was continuing development of Sirtris’ non-resveratrol synthetic selective sirtuin 1 (SIRT1) activators, which in addition to their greater potency, had more favorably drug-like properties.
Recently, resveratrol and synthetic sirtuin activators such as those developed by Sirtris have come to be known as “sirtuin-activating compounds” (STACs).
Sirtuin-activating compounds (STACs) under a cloud
As we discussed in our February 10, 2010 blog article, researchers at Amgen found evidence that the apparent in vitro activation of SIRT1 by resveratrol depended on the substrate used in the assay. The Amgen group found that the fluorescent SIRT1 peptide substrate used in the Sirtris assay is a substrate for SIRT1, but in the absence of the covalently linked fluorophore, the peptide is not a SIRT1 substrate. Resveratrol did not activate SIRT1 in vitro as determined by assays using two other non-fluorescently-labeled substrates.
Researchers at Pfizer also found that resveratrol and three of Sirtris’ second-generation STACs activated SIRT1 when a fluorophore-bearing peptide substrate was used, but were not SIRT1 activators in in vitro assays using native peptide or protein substrates.The Pfizer researchers also found that the Sirtris compounds interact directly with the fluorophore-conjugated peptide, but not with native peptide substrates.
Moreover, the Pfizer researchers were not able to replicate Sirtris’ in vivo studies of its compounds. Specifically, when the Pfizer researchers tested SRT1720 in a mouse model of obese diabetes, a 30 mg/kg dose of the compound failed to improve blood glucose levels, and the treated mice showed increased food intake and weight gain. A 100 mg/kg dose of SRT1720 was toxic, and resulted in the death of 3 out of 8 mice tested.
The Pfizer researchers also found that the Sirtris compounds interacted with an even greater number of cellular targets (including an assortment of receptors, enzymes, transporters, and ion channels) than resveratrol. For example, SRT1720 showed over 50% inhibition of 38 out of 100 targets tested, while resveratrol only inhibited 7 targets. Only one target, norepinephrine transporter, was inhibited by greater than 50% by all three Sirtris compounds and by resveratrol. Thus the Sirtris compounds have a different target selectivity profile than resveratrol, and all of these compounds exhibit promiscuous targeting.
Finally, as we reported in our December 1, 2010 blog article, NIH researcher Jay H. Chung and his colleagues found evidence that resveratrol works indirectly, via the energy sensor AMP-activated protein kinase (AMPK), to activate sirtuins. Since activation of AMPK increases fatty acid oxidation and upregulates mitochondrial biogenesis, this study suggested that the effect of resveratrol on AMPK may be more important than its more indirect activation of sirtuins in the regulation of insulin sensitivity.
All of these studies left Sirtris/GSK’s STACs under a cloud.
On March 13, 2013, GSK reported that it was shutting down Sirtris and its Cambridge MA facilities, just five years after its $720 million acquisition. GSK also said that it was offering transfers to the Philadelphia area for some of the 60 remaining Sirtris employees. Although GSK was closing Sirtris, it said that it remained confident in Sirtris’ drug candidates. The pharma company said that following Sirtris’ “highly successful” research on the biology of sirtuins, further development of Sirtris’ drug candidates “requires the resource and expertise available from our broader drug discovery organization.” GSK will be “exami[ing] [its] research against a variety of therapeutic conditions, with the aim of moving potential assets into the clinic within the next three to four years.”
New evidence that STACs activate SIRT1 in vitro under certain conditions
On 8 March 2013, the journal Science published a report by Sirtris founder David A. Sinclair, Ph.D. (Harvard Medical School, Boston MA) and his colleagues [from academia and from Sirtris, GSK, and from Biomol (Plymouth Meeting, PA)] that identified conditions under which STACs activate SIRT1 in vitro. This research report was accompanied by a Perspective in the same issue of Science authored by Hua Yuan, Ph.D. and Ronen Marmorstein, Ph.D. (Wistar Institute, Philadelphia, PA).
Dr. Sinclair and his colleagues hypothesized that the fluorophore tags on peptide substrates that were used in the original, successful SIRT1 activation assays might mimic hydrophobic amino acid residues of natural substrates at the same position as the fluorophore (i.e, +1 relative to the acetylated lysine that is engaged by SIRT1). Consistent with this hypothesis, the researchers found that non-fluorophore-tagged natural SIRT1 substrates with a large hydrophobic amino acid residue [i..e, tryotophan (Trp), tyrosine (Tyr), or phenylalanine (Phe)] at positions +1 and +6 or +1 were selectively activated by STACs. Examples of such substrates are peroxisome proliferator-activated receptor γ coactivator 1α acetylated on lysine at position 778 (PGC-1α–K778), and forkhead box protein O3a acetylated on lysine at position 290 (FOXO3a-K290). The PGC-1α–K778 peptide contains Tyr at the +1 position and Phe at the +6 position, and FOXO3a contains Trp at the +1 position. Substitution of these aromatic amino acids on either acetylated peptide with alanine (Ala) resulted in complete abolition of SIRT1 activity.
The researchers identified over 400 nuclear acetylated proteins that are potential SIRT1 targets that support STAC-mediated activation of SIRT1, on the basis of their structure. They tested five of these native sequences and found that three of them supported SIRT1 activation.
Kinetic analysis of SIRT1 activation by STACs in the presence of the above peptide substrates showed that the enhancement in the rate of SIRT1 deacetylation was mediated primarily through an improvement in peptide binding. This is consistent with an allosteric mechanism of activation. In allosteric regulation, an allosteric activator (in this case, a STAC) binds to a regulatory site (also known as an allosteric site) that is distinct from the catalytic site of an enzyme (in this case, SIRT1). Binding of the activator to the allosteric site results in the enhancement of the activity of the enzyme, for example by causing a conformational change in the protein that results in improved biding of the catalytic site to the substrate.
In order to investigate the nature of the hypothesized SIRT1 allosteric site, the researchers screened for SIRT1 mutant proteins that could not be activated by STACs in the presence of an appropriate peptide substrate. As a result of these studies, the researchers identified a critical glutamate (Glu) residue at position 230 of SIRT1, which is immediately N-terminal to the catalytic core of SIRT1. Glu230 of SIRT1 is conserved from flies to humans. Replacement of Glu230 with another amino acid, such as lysine or alanine, resulted in attenuation of SIRT1 activation by STACs, independent of the substrate used. Structural studies identified a rigid N-terminal domain that contains Glu230, and is critical for activation by STACs.
The researchers then studied the effects of STACs on cultured cells (murine myoblasts), expressing either wild-type SIRT1 or mutant SIRT1 in which Glu230 is replaced with lysine (SIRT1-E222K, which is the murine equivalent of human SIRT1-E230K). Cells expressing the mutant SIRT1 did not respond to STACs, but cells expressing wild-type SIRT1 did. Specifically, cells expressing wild-type SIRT1 exhibited STAC-stimulated increases in ATP levels, mitochondrial mass, and mitochondrial DNA copy number, but cells expressing mutant SIRT1 did not. In STAC-treated cells, the researchers found no evidence of SIRT1-independent AMPK phosphorylation. This goes against studies discussed earlier in this article, that indicate that resveratrol works via activating AMPK. They also found no evidence for inhibition of phosphodiesterase isoforms in the STAC-treated cells. This goes against a study, published in Cell in 2012, that indicates that resveratrol ameliorates aging-related metabolic conditions by inhibiting cAMP phosphodiesterases, thus engaging a pathway that activates AMPK.
The researchers conclude that STACs act via a mechanism of direct “assisted allosteric activation” mediated by the Glu230-containing N-terminal activation domain of SIRT1. They further conclude that their findings support the hypothesis that allosteric activation of SIRT1 by STACs constitutes a viable therapeutic intervention strategy for many aging-related diseases. thus apparently vindicating the Sirtris/GSK development program.
However, the authors of the companion Perspective hypothesize that the reason that existing STACs only work with SIRT1 substrates that contain hydrophobic residues at position +1 to the acetylated lysine is because they were identified via screening with a substrate that contained a hydrophobic residue mimetic–i.e., a fluorophore tag. A new screen that is not biased in this way might possibly identify STACs that exhibit selectivity for SIRT1 substrates that contain other sequence signatures. It is possible that such STACs might be better therapeutics for certain aging-related diseases than the current STACs being investigated by Sirtris/GSK. There also remain many unknowns in the biology of SIRT1, and in the biochemistry of STACs –i.e., mechanisms by with certain STACs modulate the activity of biomolecules other than SIRT1 (e.g,, cAMP phosphodiesterases). Such issues might affect the success or failure of any program to develop STACs as therapeutic compounds.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.

Pittsburgh compound B staining in AD. Source: National Institute on Aging/NIH.
In our February 28, 2013 article on the Biopharmconsortium Blog, we discussed the FDA’s February 7, 2013 Draft Guidance for Industry entitled “Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease”.
This document had been distributed for comment purposes only, and the FDA has been seeking public comment on the draft guidance for 60 days following publication.
As we discussed, by issuing this Draft Guidance, the FDA added its voice to that of an ever-increasing segment of the scientific community that calls for a new focus on conducting clinical trials in early-stage Alzheimer’s disease (AD). This is in order to focus industry R&D on developing treatments for patients whose disease is in a stage prior to the development of extensive irreversible brain damage. It is in this early stage of disease in which researchers believe that new drugs have the best chance of providing benefits to patients, by preventing further damage to the brain.
In our February 28, 2013 article, we also discussed several clinical trials being carried out by industry and academic researchers in early-stage AD. These trials should allow the scientific and medical community to answer the question as to whether treating patients with pre-AD or very early-stage AD with anti-amyloid MAb drugs can have a positive effect on the course of the disease, and slow or prevent cognitive decline.
Readers of our article may have noticed that the February 7, 2013 Draft Guidance was somewhat vague or confused. That is because there is currently no evidence-based consensus as to which biomarkers might be appropriate to support clinical findings in trials in early AD. Moreover, in “pre-AD” or very early-stage AD (i.e., before the onset of overt dementia) disease-related impairments are extremely challenging to assess accurately. Thus both measuring clinical outcomes and assessment via biomarkers in very early-stage AD are fraught with difficulty, making determination of drug efficacy very difficult.
In issuing the Draft Guidance, The FDA appeared to be seeking guidance from industry and from the academic community on how these issues might be resolved. As we said in our article, the early-stage AD trials now in progress might help the scientific and medical community, and the FDA, with issues of evaluation of biomarkers and clinical outcome measures in determining disease prognosis and the efficacy of drug treatments.
More recently–on March 13, 2013–the FDA proposed a further modification of its proposed guidelines for regulation of early-stage AD therapeutics. This was published online in an article in the New England Journal of Medicine (NEJM), entitled “Regulatory Innovation and Drug Development for Early-Stage Alzheimer’s Disease”, by Nicholas Kozauer, M.D. and Russell Katz, M.D. (As we stated in our earlier article, Dr.Katz is the director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. Dr. Kozauer is a Clinical Team Lead in the same division of the FDA.)
The new proposal attempts to deal with some of the apparent confusion in the February 7, 2013 Draft Guidance, and to facilitate the development and approval of new drugs for early-stage AD. The NEJM article notes that traditional measures of AD drug efficacy at the FDA had included assessment both of improved cognition and improvements in function. Specifically, as stated by a New York Times article discussing the new FDA proposal, “cognition” refers to such mental processes as memory and reasoning (as assessed by various tests), and “function” refers to performing such day-to-day activities as cooking, dressing or bathing.
In the FDA’s March 13, 2013 NEJM article, the authors note that researchers and regulatory agencies “simply do not yet have drug-development tools that are validated to provide measures of function in patients with Alzheimer’s disease before the onset of overt dementia”. Thus, although one can test early-stage AD patients for improvements in cognition with the appropriate tests, testing for deficits and improvements in function is extremely difficult.
The authors of the NEJM article therefore suggest that it might be feasible that a drug for treating early-stage AD be approved via the FDA’s accelerated approval pathway, on the basis of assessment of cognitive outcome alone. The agency’s accelerated-approval pathway allows drugs that address an unmet medical need to be approved on the basis of a surrogate or an intermediate clinical endpoint–in this case, a sensitive measure of improvement in cognition. Drugs approved via “accelerated approval” must be subjected to postmarketing studies to verify the clinical benefit. This regulatory pathway might facilitate the approval of treatments that appear to be effective in early AD, when patients might be expected to derive a greatest benefit than after the development of overt dementia.
With respect to selection of patients for trials in early-stage AD, the authors of the NEJM article suggest that (based on “the consensus emerging within the AD research community”) clinical diagnosis of early cognitive impairment be combined with appropriate biomarkers. These biomarkers might include brain amyloid load [as measured by positron-emission tomography (PET)] and cerebrospinal fluid levels of β-amyloid and tau proteins. The FDA places a high priority on efforts by the researchers to qualify such biomarkers in clinical trial design in early-stage AD.
The author of the New York Times article, veteran science and medicine reporter Gina Kolata, says that the FDA’s new proposal could “help millions of people at risk of developing [AD] by speeding the development and approval of drugs that might slow or prevent it.”
She also says that the proposal could be a boon for the pharmaceutical industry and AD researchers. They have often been hampered by regulations that left them uncertain of how to get drugs tested and approved for early-stage AD. Not only might anti-AD therapies provide greater benefit to patients with early-stage AD than with later stage disease, but clinical trials in early-stage AD would have a greater potential for success–provided that researchers had appropriate means of determining efficacy in early-stage AD. The new FDA proposal may increase the likelihood of identifying such appropriate means.
As pointed out in the Times article, several leading AD researchers agree, with some important caveats. For example, AD researcher P. Murali Doraiswamy, M.D. (Duke University School of Medicine) said that the new proposed regulations would lead to more clinical trials, and more motivation now to invest in the AD field. However, many companies never manage to do postmarking studies required for drugs given accelerated approval, and such studies might not be randomized clinical trials as required in gaining approval of the drugs in the first place.
Sean Bohen, M.D., Ph.D. (Senior Vice President for Early Development at Genentech) was very positive about the proposed new FDA policy, but wondered how researchers could develop appropriate tests to identify subtle cognitive changes in early AD or pre-AD. Nevertheless, he said, “We have to start somewhere.”
Thus clinical trials in early-stage AD, and development of regulatory frameworks for approval and postmarketing studies of agents that emerge from these trials, remain a work in progress.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.

Stem cells. Source: http://bit.ly/ZnYuFS
As reported in Nature News on 27 February 2013 ophthalmologist Masayo Takahashi M.D., Ph.D. and her colleagues at the RIKEN Center for Developmental Biology (Kobe, Japan), plan to submit an application to the Japanese health ministry for a clinical study of induced pluripotent stem cell (iPS)-derived cells. The researchers planned to submit their application in March 2013; if approved, they could begin recruiting patients as early as September.
The author of the Nature News article is Nature‘s Asian-Pacific Correspondent, David Cyranoski, who is based in Tokyo.
The researchers plan to treat approximately six people with severe age-related macular degeneration (AMD). Specifically, the researchers are targeting “wet” AMD, in which angiogenic blood vessels invade the retina, destroying the retinal pigment epithelium (RPE) that supports the light-sensitive photoreceptors.
AMD is a common cause of blindness that affects at least 1% of adults over 50. Wet AMD can be treated with anti-vascular endothelial growth factor (anti-VEGF) agents such as ranibizumab (Genentech/Novartis’ Lucentis), pegaptanib (Gilead/OSI/Pfizer’s Macugen), aflibercept (Sanofi/Regeneron’s Eylea), and–off-label–small doses of the anticancer agent bevacizumab (Genentech/Roche’s Avastin). However, the use of these agents requires that they be injected repeatedly into the eye.
According to the Nature News article, Dr. Takahashi and her colleagues will take an upper arm skin sample the size of a peppercorn, and transform the cells from this sample into iPS cells by using specific proteins. They will then add other factors that will induce differentiation of the iPS cells into retinal cells. Then a small sheet of these retinal cells will be placed under the damaged area of the retina, where they are expected to grow and repair the damaged RPE.
Although the researchers would like to demonstrate efficacy of this treatment in ameliorating the disease, the main focus of these studies will be on safety. Safety concerns include immunogenicity of the transplanted cells, and formation of tumors if the transplanted cells multiply uncontrollably. Another concern is that the transplanted cells might fail to engraft, and to integrate with the host tissue. It is also possible that the RPE identity of the transplanted and differentiated cells might not be stable over time.
With respect to these concerns, studies published by Japanese researchers in 2013 (Araki et al.) and reviewed in a recent Nature News article contradicted the original mouse studies that suggested that syngeneic or autologous iPS cells might be immunogenic.
With respect to tumor formation, Dr. Takahashi’s proposed studies will involve using only a few iPS cells, thus reducing the probability of forming tumors. Moreover, since the eye is relatively accessible, any tumors would be relatively easy to remove.
In addition, Dr, Takahashi has presented preclinical studies at conferences, which indicate that her iPS cells do not form tumors in mice and are safe in non-human primates. (Dr. Takahashi’s preclinical studies have also been submitted for publication.) The studies have provided reassurance of the cells’ safety to at least some leading researchers, such as Martin Pera (University of Melbourne, Australia) and George Daley (Harvard Medical School, Boston MA).
However, other researchers believe that to take iPS cell-derived tissue into the clinic at this time is premature. Robert Lanza, M.D., the chief scientific officer at Advanced Cell Technology (ACT) (Santa Monica CA) says that he cannot imagine regulatory agencies permitting studies such as Dr. Takahashi’s without years of preclinical testing.
As mentioned in the Nature News article, ACT has a program involving human embryonic stem cell (hES cell) and iPS-derived platelets for transfusion. This program is in the preclinical stage. Since platelets lack a nucleus and cannot form tumors, it is inherently less risky that clinical programs of stem-cell (and especially iPS cell) derived differentiated cells that have nuclei.
Dr. Takahashi’s proposed study of her therapy in humans is considered a “clinical study”, not a clinical trial. In Japan’s regulatory system, clinical studies are less tightly regulated than clinical trials. However, a clinical study cannot by itself lead to approval of a potential therapeutic for clinical use as a treatment. If Dr. Takahashi’s clinical study data is positive, that might attract investors or help her to get approval for a formal clinical trial. As in the U.S. or Europe, successful clinical trials will be required if Dr. Takahashi’s cellular therapy is ever to be used to treat patients.
Dr. Takahashi’s clinical study was approved by institutional review boards at both the natural sciences institute RIKEN in Wako and the Institute of Biomedical Research and Innovation in Kobe, where the surgical procedures will be carried out. Final approval will depend on the action of a committee of the Japanese Ministry of Health, Labour and Welfare. If Dr. Takahashi wins approval by September 2013 as expected, it will take another eight months to produce the tissue implants needed for her clinical study.
Other retinal repair programs involving human embryonic stem cell-derived RPE cells
Dr. Takahashi’s research does not represent the only RPE cell-based retinal repair program now being developed. There are at least two others, both of which are based on hES cells, not iPS cells.
As was not mentioned in the Nature News article, ACT has Phase 1 trials underway in its own RPE retinal repair program. ACT’s RPE cells are derived from human embryonic stem cells (hES cells). The company’s Phase 1 safety studies are in Stargardt’s Macular Dystrophy (SMD) and in dry AMD (which results from atrophy of the RPE layer, and causes vision loss through loss of photoreceptors in the central part of the eye. Dry AMD does not involve angiogenesis.). SMG is a rare inherited juvenile macular degeneration.
In February 2012, Dr. Lanza and his academic collaborators at the University of California at Los Angeles published a preliminary report of their clinical studies in dry AMD and SMG. In this study, one patient with each of the two conditions was treated with hES cell-derived RPE cells. The hES cell-derived RPE cells showed no signs of hyperproliferation, tumorigenicity, ectopic tissue formation, or apparent rejection after 4 months. Neither patient showed loss of vision, and there were signs of improvement of vision. As a result of this very preliminary study, the researchers decided in the design of future clinical studies to treat patients earlier in the disease processes, potentially increasing the likelihood of improvement of vision.
The other RPE-based retinal repair program is a collaborative effort between Neusentis (A Cambridge U.K. and Durham NC-based Pfizer research unit) and “The London Project” which was formed by Professor Pete Coffey [Institute of Ophthalmology, University College London (UCL)] and his collaborator Lyndon da Cruz (Moorfields Eye Hospital) to develop cellular therapies for all types of AMD. The London Project began collaborating with Pfizer in 2008; this collaboration was brought under the aegis of Neusentis when it was formed in 2011. Research is based on RPE cells derived from hES cells.
The Neusentis/London Project group claims to have developed a deep understanding of the biology of hEC cell-derived RPE cells, and to have worked out methods of producing enough RPE cells under GMP conditions to support clinical studies. They also claim to have developed a clear approach to establishing the safety of the therapy via preclinical studies. The collaborative group is now moving towards clinical studies of their therapies, which they “hope to achieve in the not too distant future”.
As we discussed in our February 15, 2011 article on this blog, Pfizer–as of February 1, 2011–closed its Memorial Drive laboratory in Cambridge, MA. This laboratory housed most of Pfizer’s regenerative medicine research, as well as the company’s RNAi therapeutics research group. However, as we said in this article, Pfizer was folding its Cambridge, UK regenerative medicine group–“which had been focusing on development of preclinical embryonic stem (ES) cell-based ophthalmology therapies, in collaboration with the University of London”–into a “new pain and sensory disorder research unit”. According to its website, Neusentis, which was formed in 2011, has “a particular focus on pain and sensory disorders”.
Japanese government backing for iPS cell research and commercialization
Japan has been a hotbed of iPS cell research, since these cells were first produced by Shinya Yamanaka, M.D. Ph.D. (Kyoto University) in 2006. He received The Nobel Prize in Physiology or Medicine in 2012 for his work on iPS cells. The co-recipient of the Prize, Sir John B. Gurdon, successfully cloned a frog using intact nuclei from the somatic cells of a Xenopus tadpole back in 1958. The two scientists received the 2012 Prize “for the discovery that mature cells can be reprogrammed to become pluripotent”. Since their discovery, iPS cells have been employed in such areas as basic research, disease modeling, and drug screening. (Follow this link for a recently-published example of the potential use of iPS cells in designing personalized treatments for Alzheimer’s disease.)
In 2013, as part of its stimulus package, the Japanese government has been providing generous funding for iPS research. This funding includes ¥700 million for a cell-processing centre at the Foundation for Biomedical Research and Innovation in Kobe, mainly to support Dr. Takahashi’s regenerative medicine research. In general, the iPS funding under the stimulus is aimed at moving university research on iPS cells into commercial and medical applications.
Moreover, according to Mr. Cyranoski’s 27 February 2013 Nature News article, the Japanese parliament is expected to rule by late June 2013 on a provision of a revised drug law, which would fast-track iPS-based therapies that appear to be effective in phase 2 or phase 3 trials. However, the success of the Japanese government’s efforts to accelerate commercialization of iPS-based therapies may depend in part on the success of Dr. Takahashi’s clinical research.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.
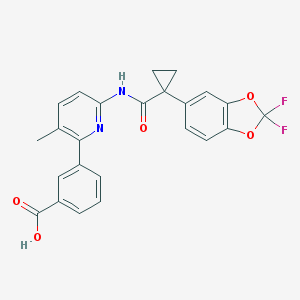
Lumacaftor (Vertex’ VX-809)
I was quoted in an article in the March 11, 2013 issue of Elsevier Business Intelligence’s The Pink Sheet by senior writer Joseph Haas. The article is entitled . A subscription is required to view the full text of this article.
The article focused on the newly-approved disease modifying drug ivacaftor (Vertex’ Kalydeco), as well as programs in drug discovery and development of disease-modifying drugs for cystic fibrosis (CF) at Vertex, PTC Therapeutics, Proteostasis Therapeutics, Pfizer, and Genzyme. It also discussed pipeline products aimed at treating or preventing life-threatening infections in CF patients at such companies as KaloBios, Insmed, and Savara.
Mr. Haas interviewed me for this article. Most of the content of our interview is available in our February 15, 2013 article on the Biopharmconsortium Blog. One company whose R&D program we did not cover in that article is Proteostasis. Proteostasis’ CF program, which is being carried out in collaboration with the Scripps Research Institute, is aimed at discovery and development of compounds that promote CFTR ΔF508 folding and trafficking. This program is in the research and lead optimization stage. We discussed R&D programs at other companies (Vertex, Pfizer) that are also aimed at correction of improper CFTR ΔF508 folding and trafficking in our February 15, 2013 article.
KaloBios’ KB001-A, a bacterial virulence factor-targeting agent
Among the agents aimed at ameliorating life-threatening infections in CF patients that were discussed in the Pink Sheet article is KB001-A, a monoclonal antibody (MAb) agent being developed by KaloBios (South San Francisco, CA). KB001-A is now in Phase 2 development for prevention of Pseudomonas aerguinosa infections in the lungs of CF patients. KB001-A targets an extracellular component of the bacterium’s type III secretion system. This system enables the bacteria to kill immune cells by injection of protein toxins into these cells.
The type III secretion system is an example of a virulence factor. Virulence factors are not expressed by a strain of pathogenic bacteria in vitro, but are expressed only when the bacteria infect a host. Once expressed, they enable the bacteria to colonize the host and cause disease.
In our June 11, 2012 article on this blog, we discussed an antibacterial drug discovery strategy aimed at targeting two related physiological systems that are important in the ability of pathogenic bacteria to cause disease, but are not essential for bacterial proliferation or survival. These systems are virulence factors and quorum sensing. At least by hypothesis, agents that disrupt these systems will prevent pathogenic bacteria from causing disease without selecting for resistant strains of the bacteria. This will give such agents an advantage over conventional antibiotics, which notoriously generate resistant strains when used to treat infections. According to the Pink Sheet article, KaloBios believes that P. aerguinosa bacteria will not develop resistance to KB001-A, which is in accord with this hypothesis.
Another issue with anti-infectives used to treat CF that is discussed in the Pink Sheet article is the definition of a “disease-modifying” agent for CF. We define disease-modifying agents as drugs that ameliorate or cure a disease by targeting the root cause of that disease. However, KaloBios considers KB001-A to be a disease-modifying agent. That is because the company believes that most CF patients die of the effects of P. aerguinosa infection, which causes deterioration of the patients’s lungs. Thus an effective anti-P. aerguinosa agent may produce dramatic increases in patients’ lifespans.
Perhaps the real issue is that one should not classify CF drugs as “disease-modifying” agent and agents that merely treat “symptoms” (as is done in the Pink Sheet article) but should define infections of CF patients as “complications” of the disease. Thus anti-infectives such as KB001-A may effectively treat a major life-threatening complication of CF, without modifying the underlying disease. Such an agent would result in increased lifespans (and improved quality of life) for CF patients, without affecting their underlying disease. As KaloBios asserts, anti-infective agents like KB001-A would be complementary to such disease-modifying agents as ivacaftor.
________________________________
As the producers of this blog, and as consultants to the biotechnology and pharmaceutical industry, Haberman Associates would like to hear from you. If you are in a biotech or pharmaceutical company, and would like a 15-20-minute, no-obligation telephone discussion of issues raised by this or other blog articles, or an initial one-to-one consultation on an issue that is key to your company’s success, please contact us by phone or e-mail. We also welcome your comments on this or any other article on this blog.